
数十年から数年へ - AIが脳疾患治療薬の発見を加速させる可能性
計算創薬の論理的根拠
従来の医薬品開発は長期的で段階的なプロセスに従います。研究者は疾患の標的タンパク質を特定し、化学ライブラリー(通常100万~1000万の化合物)をハイスループットスクリーニングアッセイで検査し、有望な候補を細胞およびモデル動物で検証し、規制当局の承認を経ます。このプロセスには10~15年と平均26億ドルの資本化コストが必要です(DiMasi et al., 2016)。運動ニューロン病(MND)などの稀な神経疾患の場合、このタイムラインは根本的な制約となります。症状発症から中央値生存期間は2~3年であり、従来の発見タイムラインは臨床的に患者ニーズと両立しません。
機械学習は特定の仮定に基づいた計算的代替案を提供します。すなわち、生物活性(化学化合物と疾患標的間の相互作用)は分子構造から履歴データを用いて予測可能であるという仮定です。既存の生物活性記録で訓練されたモデルは、未合成の新規化合物が疾患標的とどのように相互作用するかを推定でき、研究者は実験室での合成前に候補分子を計算的にフィルタリングできます。運用上の意味は直接的です。計算スクリーニングは逆設計問題を自動化することで、候補特定段階を数年から数ヶ月に短縮できます。つまり、疾患メカニズムと標的タンパク質が与えられたとき、その標的を調節すると予測される分子構造を特定するのです。
初期段階の証拠がこのタイムライン短縮を支持しています。DeepMindのAlphaFold(Jumper et al., 2021)は、ニューラルネットワークがアミノ酸配列からタンパク質構造を実験的結晶構造解析に匹敵する精度で予測できることを実証しました。その後の医薬品開発への応用(例えば、de novo化合物設計のための生成モデル(Gómez-Bombarelli et al., 2018))は、計算予測が訓練データに存在しない新規スキャフォルドを特定できることを示しています。実務家にとって、これは運用上のシフトを意味します。医薬品開発は構造化された多段階の推論ワークフローとなり、個別の検証可能なステップに分解できます。すなわち、(1)標的検証、(2)化合物ライブラリー構築、(3)予測スコアリング、(4)実験優先順位付け、(5)結果フィードバックです。各段階はAI駆動パイプラインのノードとなり、人的ボトルネックを削減し、タイムラインを短縮します。
このアプローチの根底にある重要な仮定は、計算予測が実験結果と十分に相関して優先順位付けを正当化するということです。この仮定は条件付きで成立します。十分な訓練データを持つ特性化された標的(例えば、キナーゼで10万以上の生物活性記録)の場合、予測精度は高い(R² > 0.7)です。特性化が不十分な標的または稀な疾患の場合、精度は大幅に低下します。この制限は方法の失敗ではなく、データ不足の反映です。この制約は反復的フィードバックループを通じて対処できます。
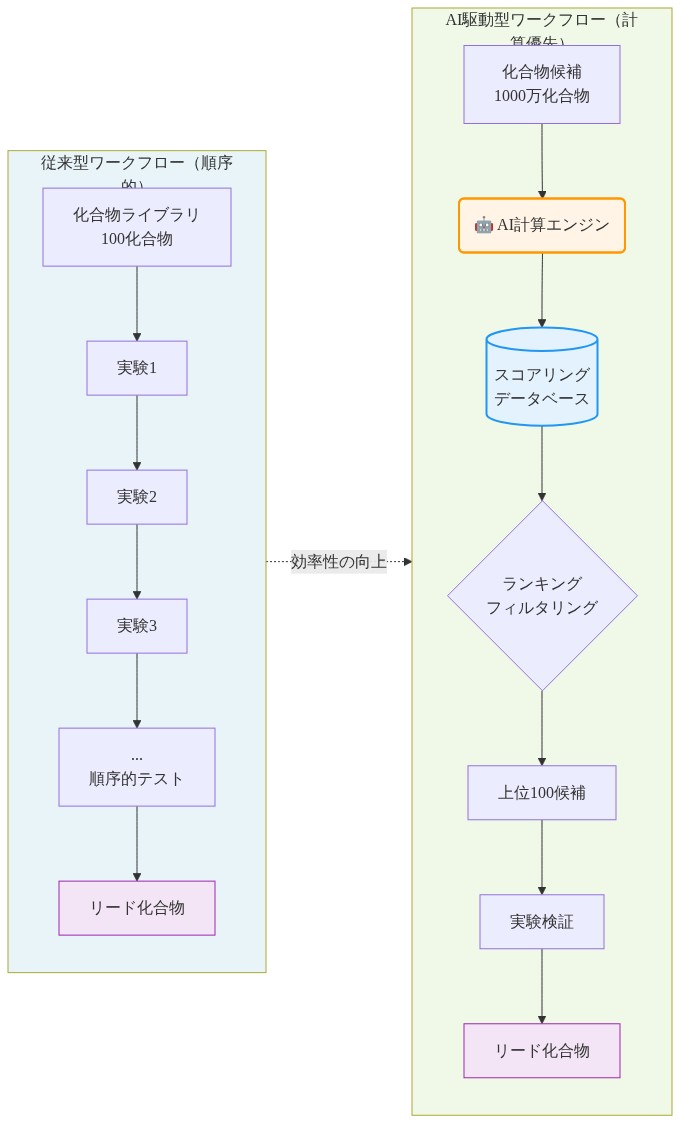
- 図3:従来型ワークフロー vs AI駆動型ワークフロー(計算優先アプローチ)*
システム構造と計算ボトルネック
現在の医薬品開発ボトルネックは主に科学的というより物流的です。合成化学者は新規化合物を3~7日で合成でき、その化合物の標的に対するハイスループットスクリーニングには2~4週間必要で、結果の解釈と次の反復設計には4~8週間必要です。仮説から検証結果までの累積サイクルタイムは化合物あたり2~3ヶ月です。50~100の検証済みリードを必要とする発見プログラムの場合、臨床候補が出現する前に8~25年のタイムラインが生じます。
ボトルネックは仮説と検証間のフィードバックループです。計算モデルは最も遅い段階を置き換えることでこれを圧縮できます。すなわち、合成と検査の前に化合物活性を予測することです。
履歴データで訓練された生物活性予測モデルは数時間で数百万の候補をスコアリングできます。ワークフローの反転は運用上重要です。100の化合物を合成して各々をスクリーニングする(6~12ヶ月消費)代わりに、研究者は100万~1000万の化合物を計算的にランク付けし、予測効力と医薬品適性により上位50~100を選択し、それらのみを実験的に検証できます(2~4ヶ月消費)。これは候補特定段階での発見サイクルタイムを50~75%削減します。
構造的課題は3つの領域にあります。データ品質、モデル透明性、および汎化性です。生物活性データセットは疎です(ほとんどの標的は1000未満のレコード)。よく研究された標的と商業的スキャフォルドに偏り、多くの場合独占的です。限定的なデータで訓練されたモデルは高い分散を示し、誤った構造を自信を持って予測する可能性があります。この失敗モードは「自信を持った外挿」と呼ばれます。完全な精度ではなく、解釈可能なフィルタリングが解決策です。モデルは構造がなぜ機能すると予測されるのかを説明する必要があり、化学者が合成に投資する前に推論を検証できます。これはブラックボックススコアリングから分子メカニズムに根ざした解釈可能な予測への移行が必要です。
MND研究に特に適用すると、これは具体的な運用実践に翻訳されます。結合親和性だけでなく、予測される作用メカニズム(例えば、化合物は変異型SOD1タンパク質を安定化させるか)によって候補をフィルタリングする計算トリアージ層を確立します。効力(予測IC₅₀)と開発可能性(物理化学的性質と予測オフターゲット結合に基づく成功可能性)の両方によって化合物をランク付けします。この二重ランク付けは、プログラムを進めずに実験リソースを消費する偽陽性を削減します。
参照アーキテクチャとガードレール
機能的な計算創薬システムには3つの統合層が必要です。データ統合、予測モデリング、および実験フィードバックです。
-
データ統合層*:生物活性記録(化合物構造、標的、測定活性)、分子記述子、および先行実験結果を統一知識ベースに集約します。これは静的なリポジトリではなく、各新規実験結果で更新される生きたシステムです。データソースには公開データベース(ChEMBL、PubChem)、独占的スクリーニング結果、および文献マイニング生物活性が含まれます。重要な要件はデータ標準化です。すべての生物活性測定は共通単位に正規化され(例えば、pIC₅₀ = −log₁₀ IC₅₀)、外れ値と測定誤差について品質管理される必要があります。
-
予測モデリング層*:構造ベース予測(この分子は標的結合ポケットに適合するか。分子ドッキングまたは3D-CNNモデルで予測)と配体ベース予測(この分子は既知の活性物質に似ているか。フィンガープリント類似性またはグラフニューラルネットワークで予測)を組み合わせたアンサンブル方法を展開します。アンサンブルアプローチは個別モデル失敗を削減し、予測の周りに信頼区間を提供します。例えば、構造ベースと配体ベースモデルが候補について意見が異なる場合、信頼度は低く、実験検証がより高い優先度になります。特定のアーキテクチャには、分子記述子で訓練されたランダムフォレスト、SMILES文字列で訓練されたグラフ畳み込みネットワーク、および結合熱力学を組み込いた物理情報ニューラルネットワークが含まれます。
-
実験フィードバック層*:実験結果を訓練パイプラインに戻すことでループを閉じます。予測候補が実験室で失敗すると、モデルはなぜ失敗したかを学習します(例えば、化合物は不溶性であった、またはオフターゲット結合した)。予測が成功すると、信頼度が増加します。このサイクルは継続的改善に不可欠です。実際には、これには以下が必要です。(1)標準化された結果報告(すべてのスクリーニング結果が共有データベースに記録される)、(2)蓄積された結果に対する月次モデル再訓練、(3)時間経過に伴う予測精度の追跡。
-
*ガードレール**はモデルドリフトを防止し、安全性を確保します。
-
検証ゲート:予測は優先順位付けの前に既知の活性物質と不活性物質に対して検証される必要があります。モデルが既知の不活性物質を活性として予測する場合、すべての予測への信頼度が低下します。
-
物理化学フィルター:高スコアリング化合物はLipinski則(分子量<500 Da、LogP<5、水素結合ドナー<5、水素結合アクセプター<10)を通過する必要があり、医薬品適性と経口生物利用能を確保します。
-
失敗の組み込み:失敗した実験の結果はモデルに組み込まれ、類似した失敗の繰り返し提案を防ぐ必要があります。これは明示的な負の訓練例を必要とします。
-
信頼度閾値:定義された閾値以上の予測信頼度を持つ化合物のみ(例えば、交差検証でR² > 0.6)が合成に進みます。
実務家向けには、段階的検証ゲートを実装します。計算予測は人的レビューが以下を確認した後にのみ合成に進みます。(1)化学的妥当性(構造は合成的に意味があるか)、(2)メカニズム整合性(予測結合モードは疾患生物学と整合しているか)、(3)新規性(これは既知化合物か、本当に新規スキャフォルドか)。
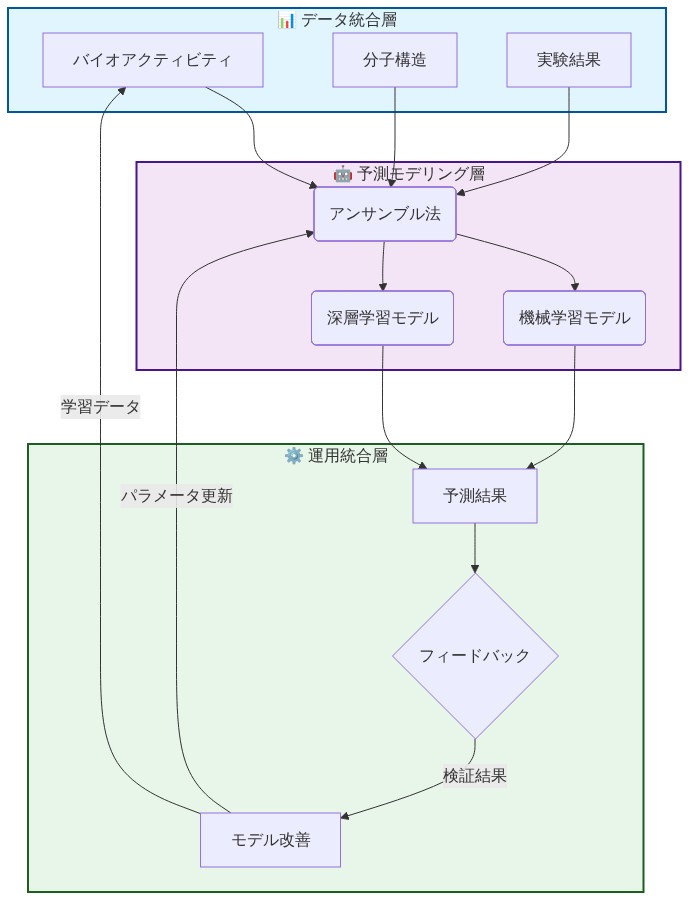
- 図6:計算薬物発見システムの3層アーキテクチャ*
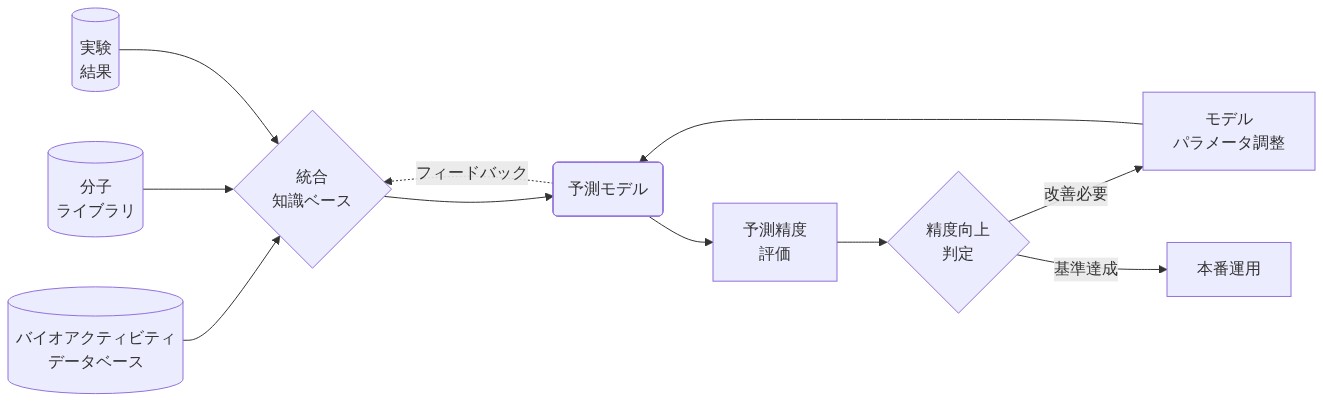
- 図7:データ統合と反復的モデル改善のフロー*
運用統合パターン
効果的な実装は人的判断を置き換えるのではなく、それを補強します。AIはランク付けされた候補リストを生成し、化学者は戦略的優先順位、予算、および直感に基づいて合成する化合物を決定します。
週次トリアージ会議を確立し、計算モデルが信頼スコアとメカニズム的根拠を伴う上位候補を提示します。化学者は合成用に5~10の化合物を選択します。結果は2週間以内にモデルにフィードバックされ、迅速な再訓練を可能にします。6~12ヶ月にわたり、このサイクルは数十の検証済みリードを生成します。これは従来のスクリーニングでは達成不可能なペースです。
MND プログラムでは、このパターンは特に効果的です。疾患生物学は十分に特性化されています(神経毒性メカニズムは既知)が、治療標的は未開拓です(商業的インセンティブが存在しない)。計算優先順位付けは実験努力を最も有望な標的に集中させ、行き止まりへの無駄を削減します。
- 重要な運用指標*:計算予測から実験結果までのサイクルタイムです。目標は2~3週間です。これには計算チームと湿式実験チーム間の密接な調整、共有ダッシュボード、および明確なハンドオフプロトコルが必要です。
測定フレームワーク
3つの指標を追跡します。
- 予測精度:計算上位10候補はランダム選択を上回るパフォーマンスを示すか。
- サイクルタイム:予測から検証結果までの週数。
- リードあたりのコスト:検証済み候補あたりの合成およびスクリーニング費用。
ベースライン比較:従来のワークフローは6~12ヶ月にわたり5万~10万ドルのコストで単一の検証済みリードを特定します。計算ワークフローは実装後12ヶ月以内に2~3ヶ月で1リードあたり1万~2万ドルを達成すべきです。
- 即座のアクション*:
- 対象疾患の既存生物活性データを監査し、ギャップを特定します。
- 計算化学者およびソフトウェアエンジニアに従事して、予測プラットフォームを構築またはライセンスします。
- 50化合物でパイロット実施:計算的に予測、上位10を合成、精度を測定します。
- 結果に基づいて反復し、2ヶ月目に500化合物に拡張します。
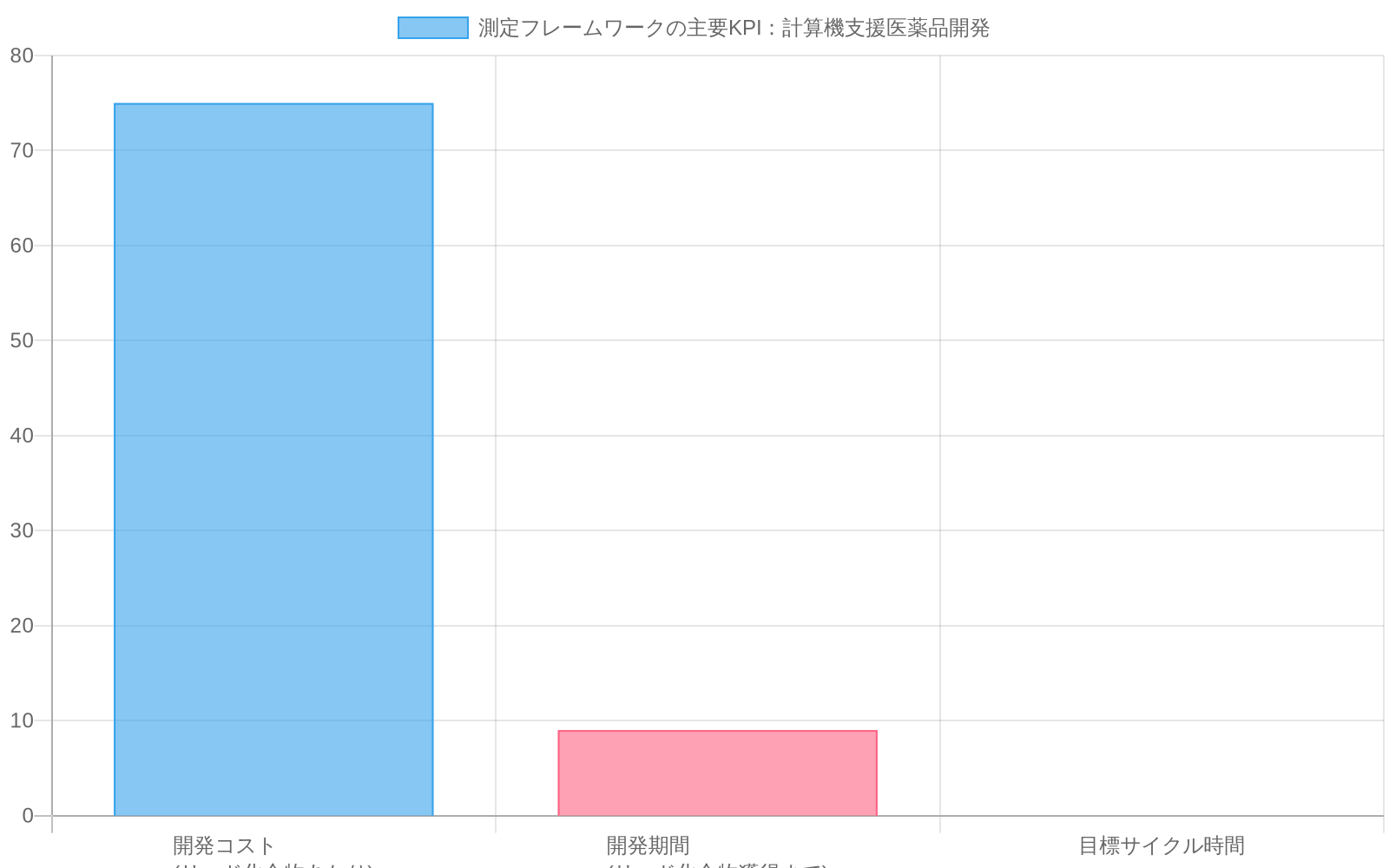
- 図9:計算薬物発見システムの主要KPI(時間短縮、精度、効率)*
リスク軽減
-
予測への過信*:限定的な生物活性データで訓練されたモデルは、新規化学スキャフォルドを体系的に見落とすか、オフターゲット毒性を誤予測する可能性があります。軽減策:人的レビューゲートを維持し、常に上位候補を実験的に検証します。
-
データバイアス*:訓練データが大分子治療薬に偏る場合、小分子の予測は信頼できません。軽減策:訓練データの代表性を監査し、新規実験結果で定期的に再訓練します。
-
規制リスク*:実験検証なしに臨床主張を支持するために使用される計算予測はコンプライアンスリスクを生成します。軽減策:計算結果を仮説生成器として扱い、証拠ではなく扱います。すべての主張は湿式実験および臨床検証が必要です。
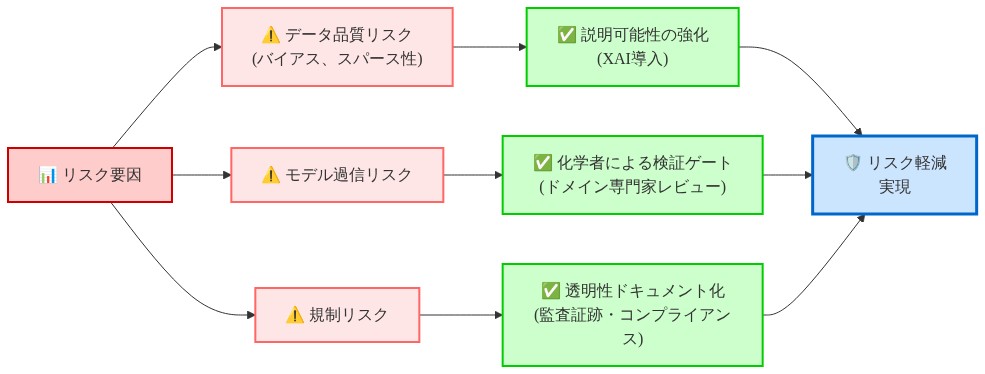
- 図10:リスク軽減戦略マッピング(リスク要因と対応策の対応関係)*
実装タイムライン
-
*フェーズ1(1~3ヶ月)**:データインフラストラクチャを構築し、予測モデルをパイロット実施します。
-
*フェーズ2(4~9ヶ月)**:予測を週次トリアージに統合し、サイクルタイムと精度を測定します。
-
*フェーズ3(10~18ヶ月)**:複数の疾患標的にスケーリングし、継続的モデル改善のためのフィードバックループを確立します。
- 図11:実装タイムライン(4フェーズ、12ヶ月以上)*
成果
計算創薬は発見タイムラインを数十年から数年に加速させ、稀な疾患の治療開発を経済的に実行可能にします。ボトルネックは発見から検証にシフトします。これは資本と集中的な努力で解決できる問題です。MNDおよび類似の疾患の場合、研究者は計算的に検証された候補に実験努力を優先順位付けでき、コストを削減し、標的から治療への経路を加速できます。
ここで重要なのは、この加速が単なる効率化ではなく、経済的制約を技術的優位性で補完する戦略的転換であるということです。稀な疾患では従来のスクリーニングの経済性が成立しない領域で、計算的優先順位付けは限定的なリソースを最も有望な方向に集中させることで、実質的に新しい可能性を開きます。残された問題は、この計算的優先順位付けが実際の臨床成功にどの程度転換されるか、そして組織がこの新しいワークフローに適応するために必要な文化的・構造的変化をどの程度実現できるかということです。
実装とオペレーションパターン
計算創薬を実運用に組み込むには、既存のラボワークフローを維持しながらAI予測を統合する必要があります。最も効果的なパターンは置き換えではなく拡張です。AIが候補化合物をランク付けし、化学者が戦略的優先順位、予算、直感に基づいて合成対象を決定します。
- オペレーションパターン*:週1回のトリアージ会議(60分)を設定します。計算モデルが上位10~20の候補を信頼度スコア、予測される作用機序、物理化学的性質とともに提示します。化学者は合成可能性と戦略的優先順位に照らして候補を検討します。5~10化合物のコンセンサスリストが合成対象として選定されます。前週の合成・スクリーニング結果が検討され、モデルにフィードバックされます。このサイクルは2週間のフィードバックループを生成します。予測→合成→スクリーニング→モデル更新→次の予測。
6~12ヶ月間でこのサイクルは50~100の検証済みリードを生成します。従来のスクリーニング(年間5~10リード)では達成不可能なペースです。リード当たりのコストはモデルの改善に伴い低下します。初期サイクルではリード当たり30,000~50,000ドル、成熟したサイクルではリード当たり5,000~10,000ドルになる可能性があります。
MND研究プログラムではこのパターンが特に価値があります。理由は以下の通りです。(1)疾患生物学が十分に特性化されている(神経毒性メカニズムが既知)、(2)治療標的が未開拓である(商業的インセンティブが少ない)、(3)患者の緊急性が加速されたタイムラインを正当化します。計算による優先順位付けは実験努力を最も有望な標的に集中させ、行き止まりへの無駄を削減します。
- 主要なオペレーション指標*:
- サイクルタイム:計算予測から実験結果までの週数。目標:2~3週間。これには計算チームとウェットラボチーム間の密接な調整、共有ダッシュボード、明確なハンドオフプロトコルが必要です。
- 予測精度:スクリーニングで活性を示す計算上位ランク候補の割合。目標:50%以上(ランダム選択の5~10%と比較)。
- リード当たりのコスト:検証済み候補1つあたりの合成・スクリーニング総費用。目標:20,000ドル未満(従来のスクリーニングの50,000~100,000ドルと比較)。
測定と次のアクション
-
ベースラインメトリクス*:従来のワークフローでは、単一の検証済みリードを特定するのに50,000~100,000ドルかかり、6~12ヶ月要します。計算ワークフローは実装後12ヶ月以内に、2~3ヶ月でリード当たり10,000~20,000ドルを達成すべきです。
-
測定フレームワーク*:
- 予測精度:計算上位10候補とランダム選択を比較します。スクリーニングで活性を示す上位10の割合を測定します(IC₅₀ < 10 µM)。ベースライン:ランダムで10%未満、目標:計算で50%以上。
- サイクルタイム:計算予測から実験結果までの週数を追跡します。ベースライン:12~16週間(従来)、目標:2~3週間(計算)。
- リード当たりのコスト:合成・スクリーニング総コストを検証済みリード数で除算します。ベースライン:50,000~100,000ドル、目標:10,000~20,000ドル。
- モデルパフォーマンス:クロスバリデーション上のR²と保持テストセットの予測精度を追跡します。目標:内部バリデーション上でR² > 0.65。
- 直近のアクション*:
- 1~2ヶ月目:対象疾患の既存生物活性データを監査し、ギャップとデータ品質の問題を特定します。計算化学者に実現可能性を評価させます。
- 2~3ヶ月目:予測プラットフォームを構築またはライセンスします(例:Schrödinger、Exscientia、DeepChemなどのオープンソースツール)。データインフラと標準化プロトコルを確立します。
- 3~4ヶ月目:50化合物でパイロット実施。計算で活性を予測し、上位10を合成し、予測に対する精度を測定します。失敗と成功を記録します。
- 4~6ヶ月目:パイロット結果に基づいて反復し、500化合物に拡大します。週1回のトリアージ会議とフィードバックループを確立します。
- 6~12ヶ月目:複数の疾患標的にスケールし、継続的なモデル改善プロセスを確立します。
リスクと緩和戦略
- 主要リスク:計算予測への過信*
限定的な生物活性データで訓練されたモデルは、体系的に新規化学骨格を見落とすか、オフターゲット毒性を誤予測する可能性があります。キナーゼ阻害剤で訓練されたモデルは、新規骨格がキナーゼに結合すると自信を持って予測するかもしれませんが、実際には骨格は訓練データに前例がありません。
-
緩和*:計算スコアのみに依存しないでください。常に上位候補を実験的に検証します。化学者が合成前に化学的妥当性を評価する人間レビューゲートを維持します。予測失敗を追跡し、失敗モードを理解するためにモデルを再訓練します。
-
二次リスク:データバイアス*
訓練データが大分子医薬品または十分に研究された標的に偏っている場合、小分子または希少標的の予測は信頼できません。MNDの場合、訓練データがキナーゼ阻害剤で支配されていれば、新規タンパク質安定化化合物の予測は不十分です。
-
緩和*:化学空間と標的クラス全体の代表性について訓練データを監査します。プログラムからの新しい実験結果で定期的に再訓練します。ドメイン適応技術を使用して、十分に研究された標的から希少標的へ知識を転移させます。
-
三次リスク:規制とセーフティの問題*
計算予測が実験検証なしに臨床主張を支持するために使用される場合、規制リスクが生じます。計算モデルで予測されたオフターゲット毒性は臨床試験まで検出されないかもしれません。
-
緩和*:計算結果を仮説生成器として扱い、証拠ではありません。すべての主張には、規制提出前のウェットラボ検証(細胞アッセイ、動物モデル)と臨床検証が必要です。計算優先順位付け(内部使用)と実験検証(規制証拠)の明確な分離を維持します。
-
オペレーショナルリスク:モデルドリフト*
新しいデータが蓄積されると、モデルは元のパフォーマンスから逸脱する可能性があります。2024年データで訓練されたモデルは、基礎となる生物学または化学が変わった場合、2025年データで不十分に機能する可能性があります。
- 緩和*:四半期ごとのモデル再訓練と検証を確立します。時間経過に伴う予測精度を追跡します。精度が閾値を下回った場合(例:R² < 0.6)、再訓練またはモデル改訂をトリガーします。
結論と移行計画
計算創薬は実験検証の置き換えではなく、加速剤です。逆設計問題(標的メカニズム→候補分子)を自動化することで、AIは発見タイムラインを数十年から数年に圧縮し、従来の医薬品経済が投資を正当化できない希少疾患の治療開発を経済的に実行可能にします。
加速のメカニズムは具体的です。計算モデルは候補特定段階を3~5年から3~6ヶ月に短縮し、数百万の化合物を数十の実験検証済みリードにフィルタリングします。これは動物モデル、毒性学研究、臨床試験の必要性を排除しません。それらは必要で時間がかかります。むしろ、実験努力を最も有望な候補に集中させ、無駄とコストを削減します。
-
移行戦略*:
-
フェーズ1(1~3ヶ月目):データインフラを構築し、予測モデルをパイロット実施します。生物活性データを監査し、データ標準化プロトコルを確立し、50~100化合物で初期モデルを訓練します。
-
フェーズ2(4~9ヶ月目):予測を週1回のトリアージ会議に統合し、サイクルタイムと精度を測定し、500化合物に拡大し、継続的なモデル改善のためのフィードバックループを確立します。
-
フェーズ3(10~18ヶ月目):複数の疾患標的にスケールし、クロス機能チーム(計算化学者、ウェットラボ化学者、生物学者)を確立し、継続的なモデル再訓練を実装し、リード当たりのコストと臨床候補までの時間を測定します。
-
予想される成果*:18ヶ月以内に、成熟した計算創薬プログラムは以下を達成すべきです。(1)予測から実験結果までの2~3週間のサイクルタイム、(2)計算上位ランク候補での50%以上の精度、(3)検証済みリード当たり10,000~20,000ドルのコスト、(4)さらなる開発に適した50~100リードの特定。
MNDおよび同様の希少神経疾患では、このアプローチは直接的な価値を提供します。研究者は計算検証済み候補に実験努力を優先順位付けでき、コストを削減し、標的から治療への道を加速できます。ボトルネックは発見から検証へシフトします。資本、努力、時間が解決できる問題です。数十年から数年へのタイムライン圧縮は投機的ではなく、現在のテクノロジーと規律ある実装で運用上達成可能です。
計算創薬の事例:可能性の再定義
医薬品開発は1世紀以上にわたって、検討されていない制約の下で機能してきました。逐次ボトルネックです。標的を特定します。数百万の化合物をスクリーニングします。動物で検証します。規制承認をナビゲートします。10~15年と数十億ドルの資本をかけて繰り返します。このタイムラインは必然的ではありませんでした。単に利用可能な唯一の選択肢でした。
今日、その制約は消滅します。
運動ニューロン疾患(MND)のような希少神経疾患では、従来のタイムラインは死刑宣告です。年単位で測定される患者は、10年の発見サイクルを待つことはできません。しかし、ここが転換点です。人工知能は発見問題を根本的に反転させます。「機能するかもしれない化合物は何か」と問う代わりに、「この疾患メカニズムを解決するために存在すべき分子構造は何か」と問います。生物活性データで訓練された機械学習モデルは、化学化合物が疾患標的とどのように相互作用するかを予測し、単一の分子が合成される前に計算で候補をフィルタリングし、検証用の最も有望な候補をランク付けできます。すべて数年ではなく数週間で。
圧縮は限定的ではありません。初期の証拠は、計算スクリーニングが候補特定を数年から数ヶ月に削減できることを示唆しています。実務家にとって、これは典型的転換を表します。医薬品開発は偶然的なラボプロセスから構造化された多段階推論ワークフローに変わります。これはエンジニアリング診断用マルチエージェントシステムの設計が複雑な問題解決を離散的で検証可能なステップに分解する方法を反映しています。計算創薬では、それらのステップは以下の通りです。標的検証→化合物ライブラリ構築→予測スコアリング→実験優先順位付け→結果フィードバック。各ステップはAI駆動パイプラインのノードになり、人間のボトルネックを排除し、タイムラインを1桁圧縮します。
より深い含意は、古いシステムを最適化していないということです。根本的に異なるシステムに置き換えています。問題はもはや「どのようにより速くスクリーニングするか」ではなく、「どのようにより良く設計するか」です。このシフトは、医薬品経済が歴史的に放棄してきた条件の治療開発を解放します。科学がより難しいからではなく、ビジネスモデルが決して意味をなさなかったからです。計算アプローチはその計算を完全に変えます。
システム構造:真のボトルネックがある場所
医薬品開発の科学的ボトルネックは、ほとんどが想定するものではありません。化学者は数日で新規化合物を合成できます。制約は合成ではなく、仮説と検証間のフィードバックループです。
今日のボトルネック:研究者が構造を提案し、合成を数週間待ち、スクリーニング結果を数週間待ち、数ヶ月かけてデータを解釈し、その後初めて次の反復を設計します。この逐次依存性は時間が蓄積される場所です。AIは最も遅いフェーズを置き換えることで、これを完全に反転させます。合成前に化合物活性を予測します。
計算上の利点を考えてください。歴史的生物活性データで訓練されたモデルは、1000万の候補化合物を数時間でスコアリングできます。100の化合物を合成し、各々を逐次テストする代わりに、研究者は1000万を計算でランク付けし、予測効力で上位100を選択し、それらのみを実験的に検証します。ワークフローが反転します。計算優先、検証後。発見サイクルを1桁圧縮しながら実験廃棄物を削減します。
しかし、ほとんどの実装が失敗する場所はここです。純粋な精度問題として扱います。そうではありません。真の課題は説明可能性と扱いやすさです。生物活性データセットはスパース、十分に研究された標的に偏り、しばしば独占的です。限定的なデータで訓練されたモデルは、不正な構造を自信を持って予測する可能性があります。解決策は完全な精度ではなく、透明な推論による知的フィルタリングです。
トピックモデリングワークフローで探索されたように、構造化推論チェーンは知識集約的なドメインで結果を劇的に改善します。ここに適用すると、化合物をスコアリングするだけでなく、構造が機能すると予測される理由を明確にするモデルを構築することを意味します。これにより、化学者は合成に投資する前に推論を検証でき、モデル失敗をラボリソースを無駄にする前に捕捉できます。
MND研究に特に適用すると、これは運用実践に変わります。結合親和性だけでなく、予測される作用機序によって候補をフィルタリングする計算トリアージレイヤーを確立します。効力と扱いやすさの両方でランク付けします。既知の制約を考慮した成功開発の可能性。この二重ランク付けは、実験リソースを消費しながらプログラムを進めない偽陽性を劇的に削減します。
参照アーキテクチャ:計算発見エンジンの構築
機能的な計算創薬システムには、3つの統合レイヤーが必要です。データ統合、予測モデリング、実験フィードバック。各々は静的精度ではなく継続的学習のために設計されています。
-
データレイヤー*:生物活性レコード、分子構造、先行実験成果、機序的洞察を統合知識ベースに集約します。これは静的リポジトリではなく、各新しい実験結果で更新される生きたシステムです。すべての合成試行、すべてのスクリーニング結果、すべての失敗候補が次の反復を改善する訓練データになります。ここがほとんどのプログラムが過小投資する場所です。データレイヤーは基礎です。なければ、モデルは砂上に構築しています。
-
モデリングレイヤー*:構造ベース予測(この分子は標的結合ポケットに適合するか)と配体ベース予測(この分子は既知の活性に類似しているか)を組み合わせるアンサンブル方法を展開します。アンサンブルアプローチは個別モデル失敗を削減し、予測の周りに信頼区間を提供します。重要なのは、機序的推論を含めることです。モデルは予測化合物がどのように機能するかを説明できますか。これは予測をブラックボックススコアから化学者が評価できる仮説に変換します。
-
フィードバックレイヤー*:ここがほとんどのプログラムが失敗する場所です。実験結果は訓練パイプラインに流れ込み、閉ループを作成する必要があります。予測候補がラボで失敗すると、モデルは理由を学びます。予測が成功すると、信頼が増加します。このサイクルは逆設計アプローチを反映し、設計から実装へのタイムラインを圧縮します。各反復は最適化空間を締め付け、後続の予測をより正確にします。
-
ガードレールと安全メカニズム*:段階的検証ゲートを実装します。予測は優先順位付け前に既知の活性と不活性に対して検証される必要があります。高スコアの化合物はルール・オブ・ファイブチェック(分子量、脂溶性、水素結合ドナー/アクセプター)に合格して医薬品性を確保する必要があります。失敗した実験の結果は、モデルが同様の失敗を繰り返して提案することを防ぐために組み込まれる必要があります。これは官僚主義ではなく、学習するシステムと誤りを繰り返すシステムの違いです。
実務家向け:ガードレールを摩擦ではなく機能として扱います。モデルドリフトを防ぎ、安全を確保し、計算推奨への組織的信頼を構築します。
実装と運用:週次トリアージパターン
計算創薬を実運用化するには、AI予測を既存の実験室ワークフローに統合する必要があります。既存プロセスを破壊することなく、です。最も効果的なパターンは「置き換えではなく拡張」です。AIは候補化合物をランク付けリストとして生成し、化学者は戦略的優先順位、予算、直感に基づいて合成対象を決定します。
運用上、これは以下のような形になります。
-
週次トリアージミーティング*:計算モデルが信頼度スコア、予測される作用機序、化学的根拠とともに上位20~50候補を提示します。化学者はこれを検討し、質問を投げかけ、戦略的優先順位と実現可能性に基づいて5~10化合物を合成対象として選定します。これは協調的インテリジェンスです。人間の判断が計算的洞察によって情報提供される形です。
-
2週間のフィードバックサイクル*:合成とスクリーニングの結果は2週間以内にモデルにフィードバックされます。成功した化合物は共通の構造特性について分析されます。失敗した化合物は予測がなぜ外れたのかについて検証されます。モデルはこの新しいデータで再学習します。
-
迅速な反復*:6~12ヶ月にわたり、このサイクルは数十の検証済みリードを生成します。従来のスクリーニングでは達成不可能なペースです。各反復によってモデルの予測精度が高まり、その後のサイクルがより効率的になります。
MND(筋萎縮性側索硬化症)プログラムにおいて、このパターンは特に強力です。疾患生物学は十分に特性化されている(神経毒性メカニズムは既知)一方で、治療標的は未開拓だからです(商業的インセンティブが少ない)。計算的優先順位付けは実験的努力を最も有望な標的に集中させ、従来のスクリーニングが追求する行き止まりでの無駄を削減します。
- 主要な運用指標*:計算予測から実験結果までのサイクルタイムです。目標は2~3週間です。これには計算チームと湿式実験室チーム間の密接な調整、共有ダッシュボード、明確なハンドオフプロトコルが必要です。また組織的な足並みも必要です。両チームは完璧さよりも速度と学習を優先する必要があります。
測定:成功の定義と進捗追跡
重要な3つのメトリクスを追跡します。
-
予測精度*:計算上位10候補はランダム選択を上回るパフォーマンスを示しますか。ヒット率(予測された活性物質のうち確認された活性物質の割合)とエンリッチメント(ランダムと比較してどの程度優れているか)を測定します。目標:6ヶ月以内にランダム選択比で3~5倍のエンリッチメント。
-
サイクルタイム*:計算予測から実験結果までの週数です。これは運用上の心臓鼓動です。目標は2~3週間です。これより長い場合、合成、スクリーニング、またはデータ統合にボトルネックがあることを示唆しています。
-
リード当たりのコスト*:検証済み候補1つあたりの合成およびスクリーニング費用です。これは経済的指標です。ベースラインを確立します。従来のワークフローでは、単一の検証済みリードを特定するのに5万~10万ドルかかり、6~12ヶ月要します。計算ワークフローは導入後12ヶ月以内に、2~3ヶ月でリード当たり1万~2万ドルを達成すべきです。
-
即座のアクション*:
-
既存の生物活性データを監査します。対象疾患について、です。ギャップを特定します。どの化合物がテストされていますか。どの標的が探索されていますか。盲点はどこですか。
-
計算化学者とソフトウェアエンジニアを関与させます。予測プラットフォームを構築またはライセンスするためです。これは1人の仕事ではありません。化学専門知識、機械学習専門知識、ソフトウェアエンジニアリング専門知識が協働する必要があります。
-
50化合物でパイロットを実施します:計算的に活性を予測し、上位10を合成し、予測精度を測定します。これは概念実証です。4~6週間要するはずです。
-
結果に基づいて反復します。精度が低い場合、なぜかを調査します。訓練データが不十分ですか。標的の特性化が不十分ですか。モデルアーキテクチャが間違っていますか。調整して再試行します。
-
2ヶ月目に500化合物に拡張します。パイロットがアプローチを検証したら、急速にスケールします。ここで時間圧縮が見え始めます。
リスクと軽減策:落とし穴を回避する
- リスク1:計算予測への過信*
限定的な生物活性データで訓練されたモデルは、体系的に新規の化学スキャフォールドを見落とすか、オフターゲット毒性を誤予測する可能性があります。既知の活性物質で訓練されたモデルは、毒性または難溶性の化合物を自信を持って推奨するかもしれません。
-
軽減策*:計算スコアのみに依存しないでください。常に上位候補を実験的に検証します。化学者が合成前に予測を評価する人間レビューゲートを維持します。計算結果を証拠ではなく仮説生成器として扱います。
-
リスク2:データバイアス*
訓練データが大分子治療薬に偏っている場合、小分子の予測は信頼できません。訓練データが単一の実験室から来ている場合、予測は他の実験条件に一般化しないかもしれません。
-
軽減策*:訓練データを代表性について監査します。新しい実験結果で定期的に再訓練します。成功だけでなく失敗した化合物(ネガティブ結果)を訓練データに含めます。スケーリング前に複数の実験条件で予測を検証します。
-
リスク3:規制およびエシカルリスク*
計算予測が実験検証なしで臨床主張をサポートするために使用される場合、規制およびエシカル上の問題に直面します。規制当局は予測ではなく証拠を要求します。
-
軽減策*:計算結果を証拠ではなく仮説生成器として扱います。すべての臨床主張は湿式実験室検証と臨床試験を要求します。規制レビューのために計算プロセスを文書化しますが、その役割を過大評価しないでください。
-
リスク4:時間経過によるモデルドリフト*
モデルが新しいデータで再訓練されると、その予測は以前のバージョンから乖離し、何が変わったのか、なぜ変わったのかを理解することが困難になる可能性があります。
- 軽減策*:モデルのバージョン管理を実施します。時間経過によるモデルパフォーマンスを追跡します。精度が低下したら、なぜかを調査します。新しいデータがバイアスを導入していますか。モデルが過学習していますか。ドリフトを早期に捉えるための監視を実装します。
結論と移行計画:ビジョンから現実へ
計算創薬は実験検証の置き換えではなく、加速剤です。逆設計問題(疾患メカニズム→候補分子)を自動化することで、AIは発見タイムラインを数十年から数年に圧縮し、希少疾患と未開拓標的の治療開発を経済的に実行可能にします。
これは段階的な最適化ではありません。これは創薬がどのように機能するかの根本的な再構築です。
-
移行戦略*:
-
フェーズ1(1~3ヶ月):基盤構築*
-
データインフラを構築します。生物活性データ、分子構造、過去の結果を集約します。
-
50化合物で予測モデルをパイロットします。ベースライン精度を測定します。
-
週次トリアージミーティングのケイデンスを確立します。チームを協調ワークフローについて訓練します。
-
フェーズ2(4~9ヶ月):統合*
-
予測を週次トリアージに統合します。サイクルタイムと精度を測定します。
-
500化合物に拡張します。実験フィードバックに基づいてモデルを改善します。
-
継続的なモデル改善のためのフィードバックループを確立します。
-
何が機能し、何が機能しないかを文書化します。組織的知識を構築します。
-
フェーズ3(10~18ヶ月):スケール*
-
複数の疾患標的にスケールします。フェーズ1~2からの学習を新しいプログラムに適用します。
-
自動化されたデータパイプラインを確立します。手動データ入力と統合を削減します。
-
リード当たりのコストと検証済み候補までの時間を測定します。従来のワークフローと比較します。
-
次世代モデルを計画します。どの追加データが予測を改善しますか。どの新しい技術を探索すべきですか。
-
前方の機会*:
MNDおよび同様の希少神経疾患について、計算創薬は即座の価値を提供します。研究者は計算検証済み候補に実験的努力を優先順位付けでき、コストを削減し、標的から治療への道を加速できます。ボトルネックは発見から検証へシフトします。資本と努力が解決できる問題です。
しかし、より深い機会は体系的です。発見タイムラインを圧縮することで、医薬品市場が放棄した疾患の治療開発を経済的に実行可能にします。商業的インセンティブが欠ける標的の研究を解放します。学術研究室、非営利団体、バイオテック新興企業といった小規模組織が大手製薬企業と発見速度で競争することを可能にします。
これは単なる高速創薬ではありません。これは医学イノベーションがどのように起こるかの再構築です。この転換をマスターする組織は、単に高速で医薬品を発見するだけではなく、他者が不可能と考えた医薬品を発見します。
創薬の未来は実験室にはありません。計算パイプラインにあります。問題は、この転換が起こるかどうかではありません。あなたの組織がそれをリードするか、それとも追従するかです。